
Virtual Cell Challenge Shenanigans
Hacking the DES metric and some thoughts


When my classmates and I decided to team up to try our hand at the Virtual Cell Challenge [1], we were initially excited to explore ML techniques to predict unseen perturbations. Over the summer, we brainstormed lots of ideas, particularly in learning robust log fold change estimates and gene dependencies.
However, digging around the VCC training data we found some features we could potentially exploit to hack some of the metrics, particularly the DES score. So eventually we went the hacking route. While other teams evidently had much more sophisticated forms of hacking and actually approaching the problem, we would like to share our experience messing around, what we found, and some of our takeaways.
The following assumes the reader was an active participant in the VCC, or moderately aware of how the challenge is structured.
Predicting number of DEGs from given data
The challenge provided the number of cells and median number of UMIs. Part of the variability across perturbations in these values is likely correlated with the degree to which a perturbation affects the cell (i.e. cell viability). In fact, we see a clear relationship between nCells x nUMIs and the number of DEGs (differentially expressed gene, calculated by cell-eval, fdr < 0.05).
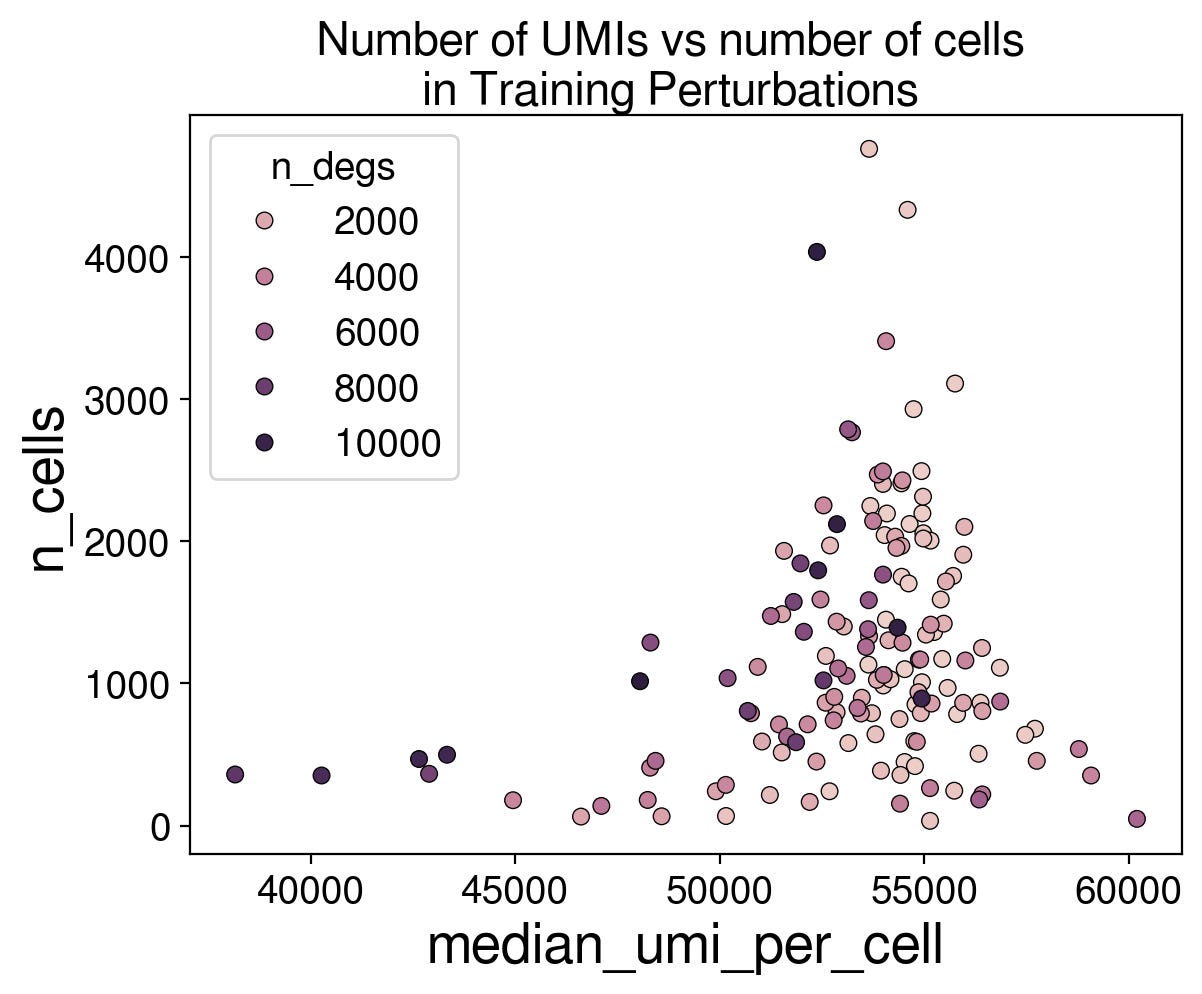
To see if this information could be used to gain performance on any of the metrics, I used nCell and nUMI to query the nearest training data perturbation (using Mahalanobis distance), and then sampled the transcriptional profiles of the cells from that perturbation.
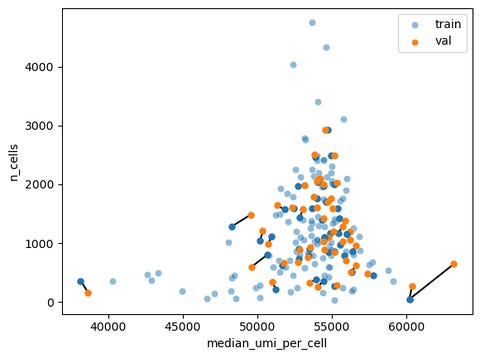
This approach gave us 9.4 (0.18/0.61/0.036) on the validation set, though soon after it was bumped down to 5.8 (0.18/0.56/0.036) with the bug fix in the PDS score calculation at the beginning of October. This gain in performance is largely driven by predicting which perturbations have no effect. Perturbations that minimally affect the cells will likely have an nCell x nUMI similar to each other and to non-targeting controls.
To improve on this, we could first predict the number of DEGs based on nCells and nUMIs with linear regression with an interaction term.
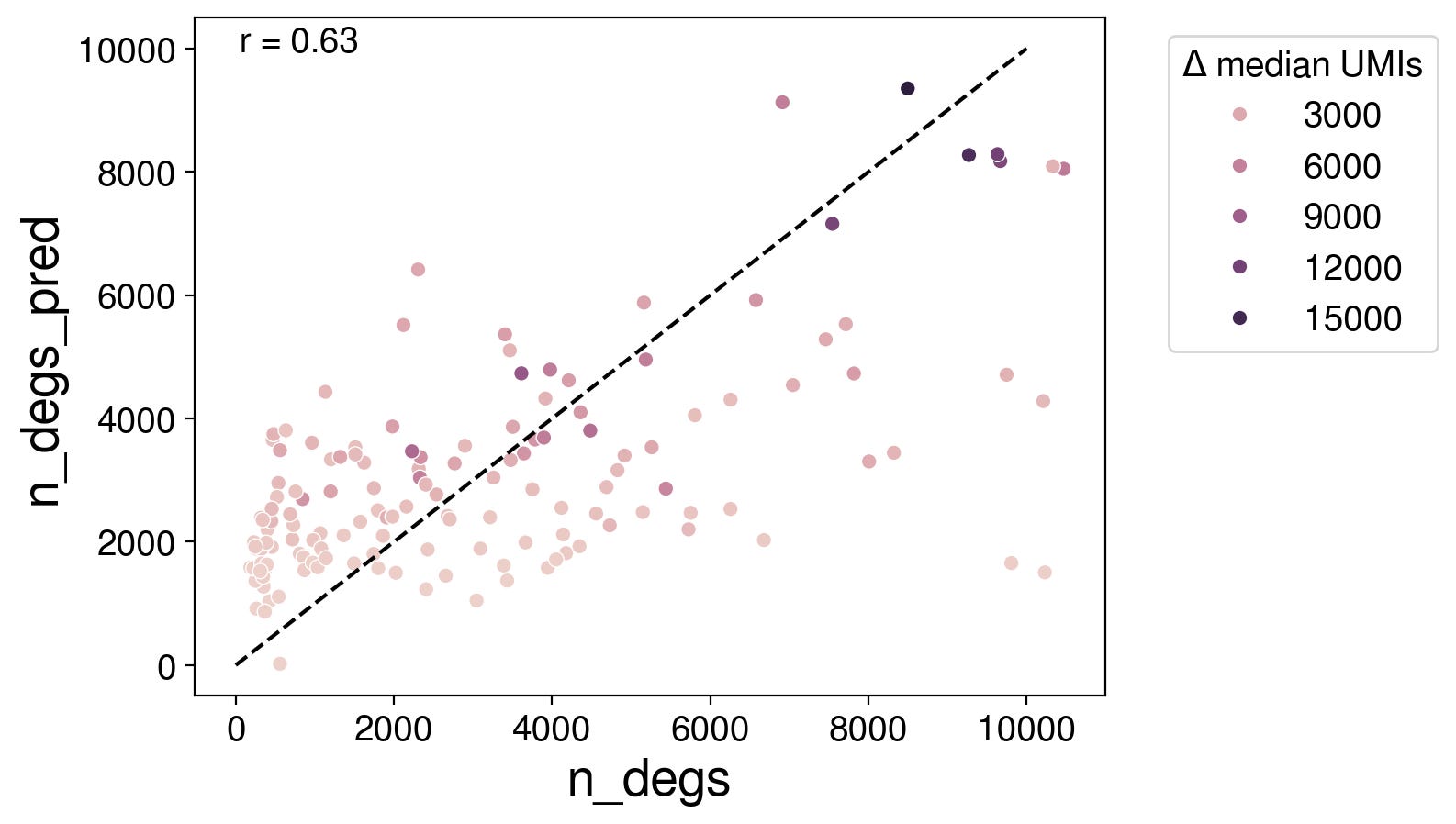
Now, we just need to find a rational strategy to select which genes to be DEGs.
DEGs are often highly expressed genes in control cells
Looking more closely at the “true” DEGs in the training data, we found that many genes were frequently DEGs, even up to ~100 out of the 150 training perturbations.
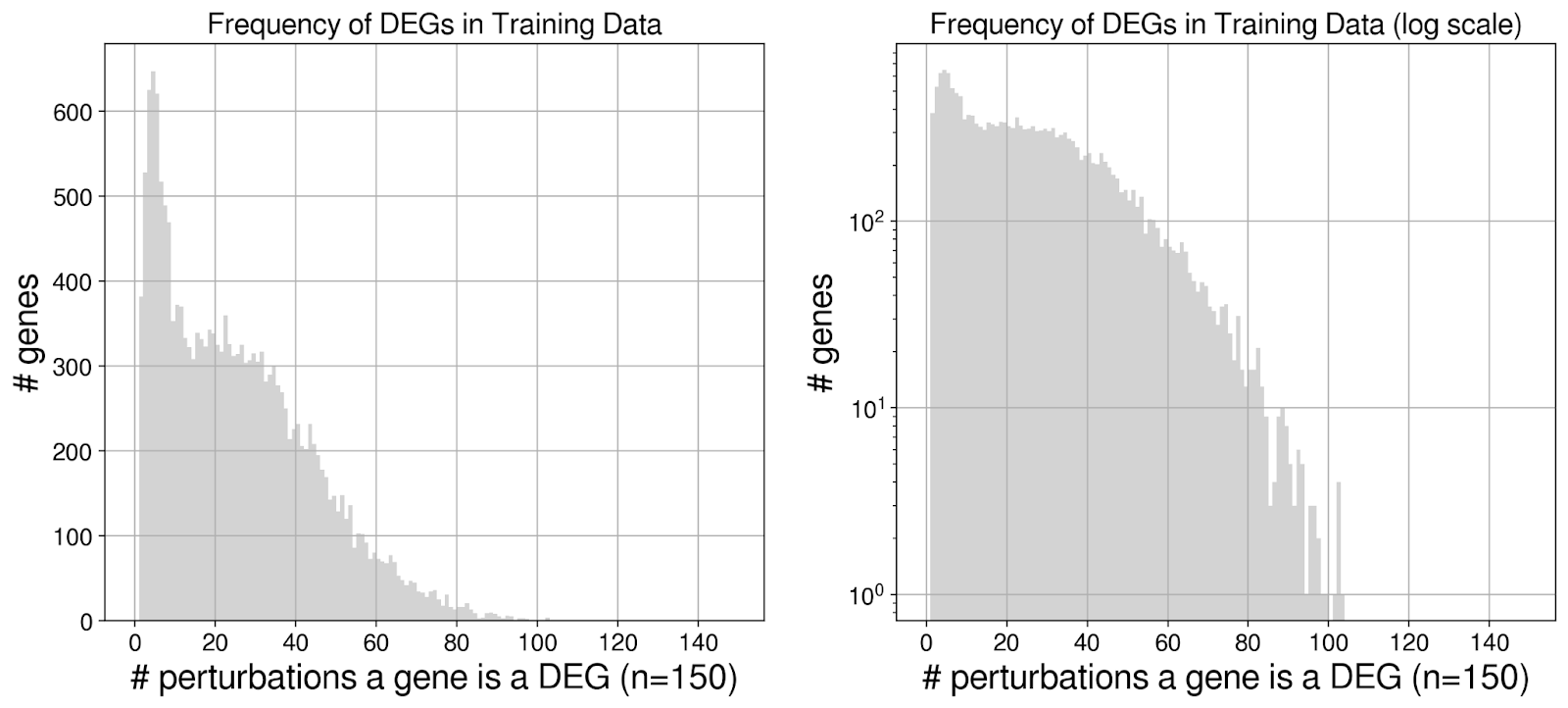
What made some genes more likely to be a DEG? Turns out it is highly correlated with the mean expression in the non-targeting control cells.
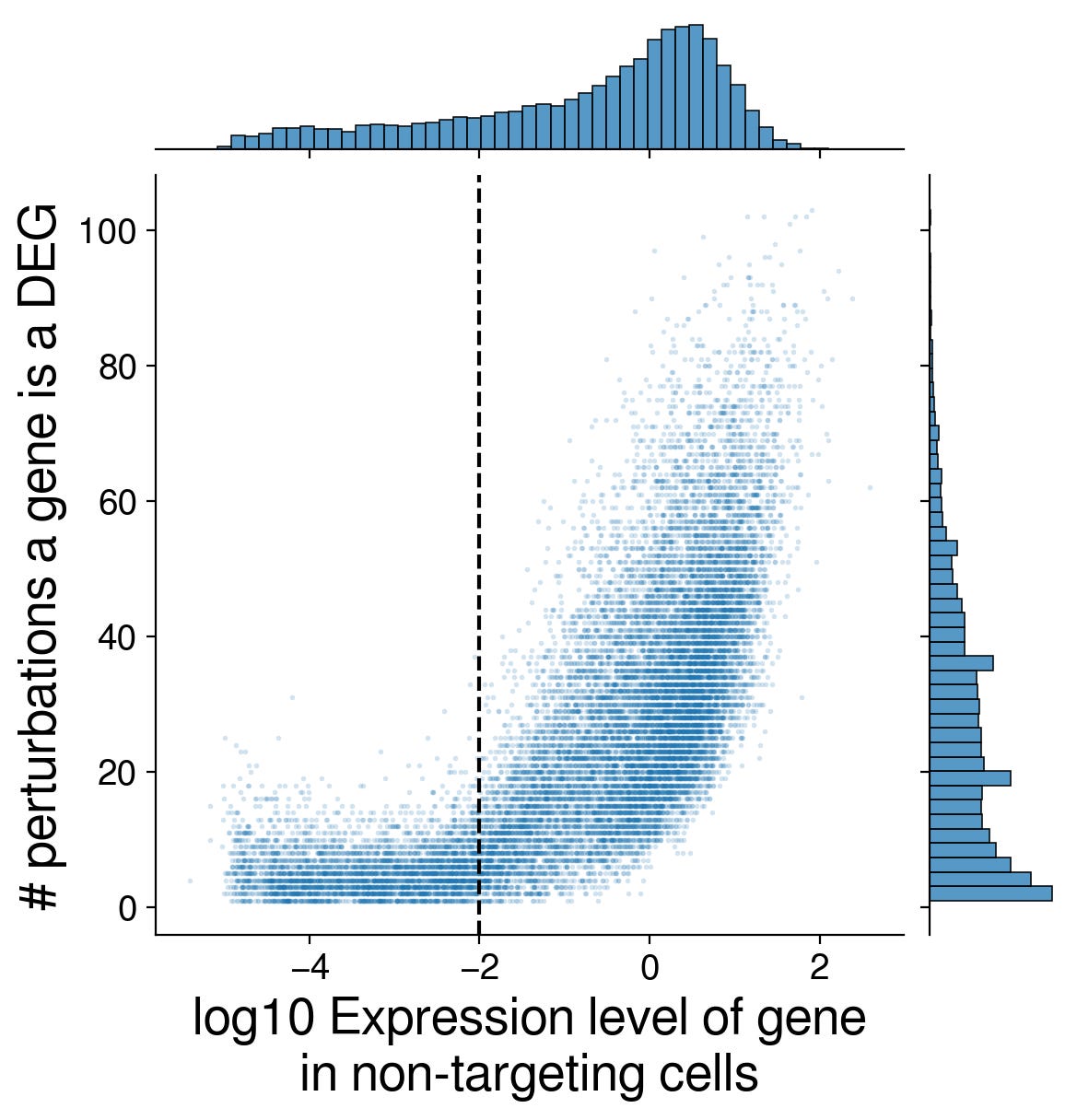
Furthermore, we found that as a perturbation had more DEGs, the DEGs were approximately prioritized almost in rank order of their non-targeting control expression level.
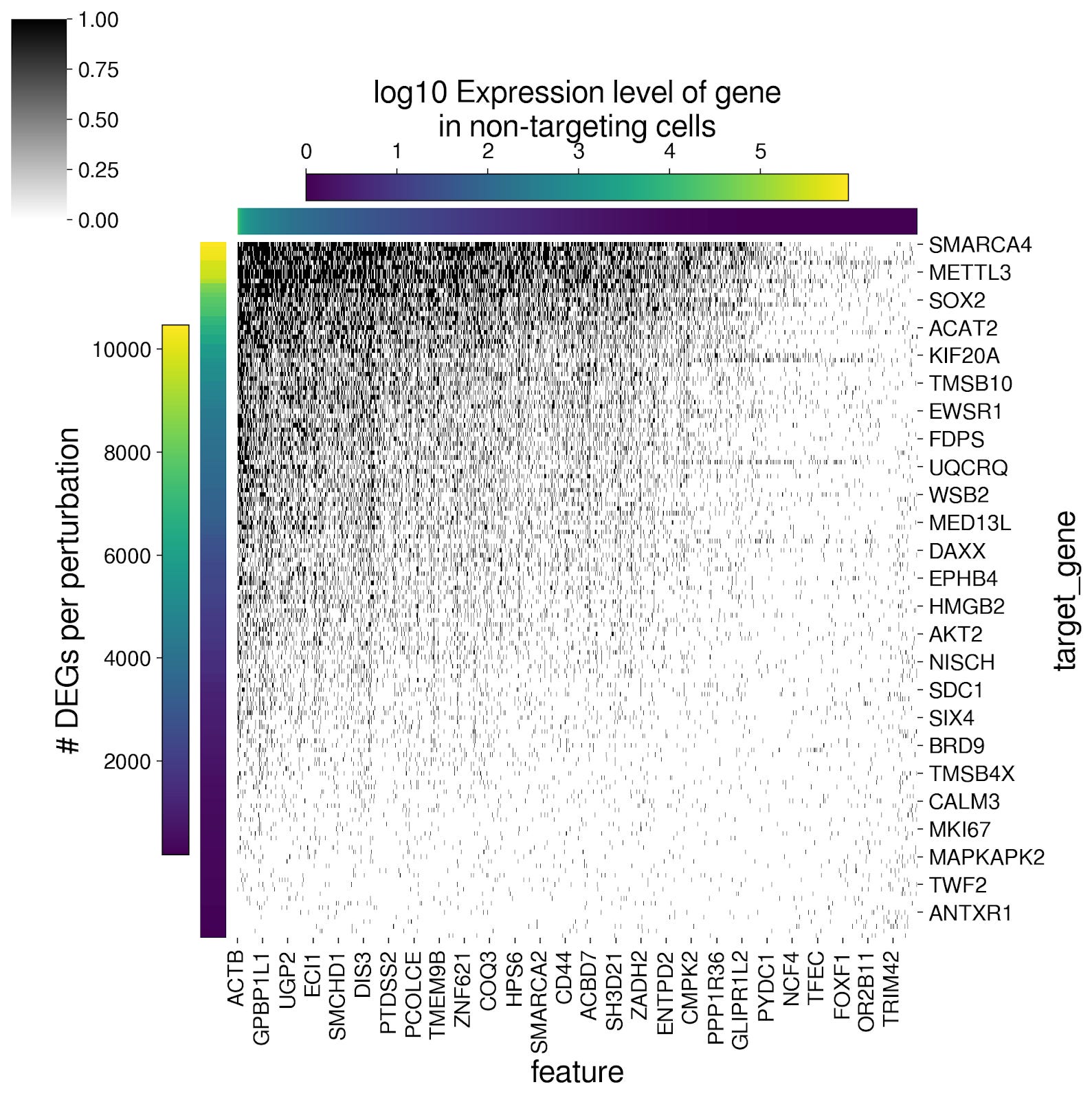
So simple approach to maximize the DES score could be:
Predict the number of DEGs K using nCells and nUMIs with simple linear regression
Given all 18k genes ranked in descending order by expression level in the non-targeting controls, take the top K genes.
Make these K genes DEGs, and keep everything else the same as the non-targeting control cells.
To make these K genes a DEG, I predicted the log fold change (LFC) from the training data. Generally, the frequency a gene is a DEG (which is approximately the same as its non-targeting control expression level) is inversely proportional to the LFC. This makes sense since it is hard to jump orders of magnitudes for a gene that is already highly expressed.
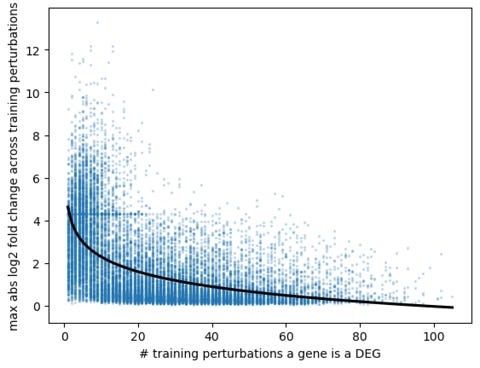
Using this approach, my overall score was 5.3 (0.239/0.519/0.157). The overall score was not better than the nCellxnUMI nearest neighbor approach (5.8) even though my DES score was higher at 0.239. This is because I did not get any improvement on the PDS score compared to baseline.
A variant on the transformation hack
It was around mid-October that discussions about data transformations on discord came to my attention (Arc Virtual Cell Challenge part 2: What the Leaderboard Collapse Reveals About Model Evaluation) [2].
From what I could understand from the online discussions, some teams were able to inflate their PDS and DES scores alone by submitting their control cells as raw counts and their predicted perturbed cells as log1p normalized cells. While I did not know what was happening with the PDS score (see a cool analysis on the PDS score by Liu et al. [3]), the effect on the DES score made sense.
First, such a difference in normalization (and lack of) must make all genes a DEG. The distributions of raw counts and log1p counts are just so different. Second, if your predictions produce more DEGs than the “true” number K, the DES only looks at the top K predicted DEGs by highest LFC. High expressed genes would get the highest LFC since they get compressed the most with log1p normalization. Below, I computed the LFC between log1p normalized counts and raw counts of just the non-targeting control cells to demonstrate this effect.
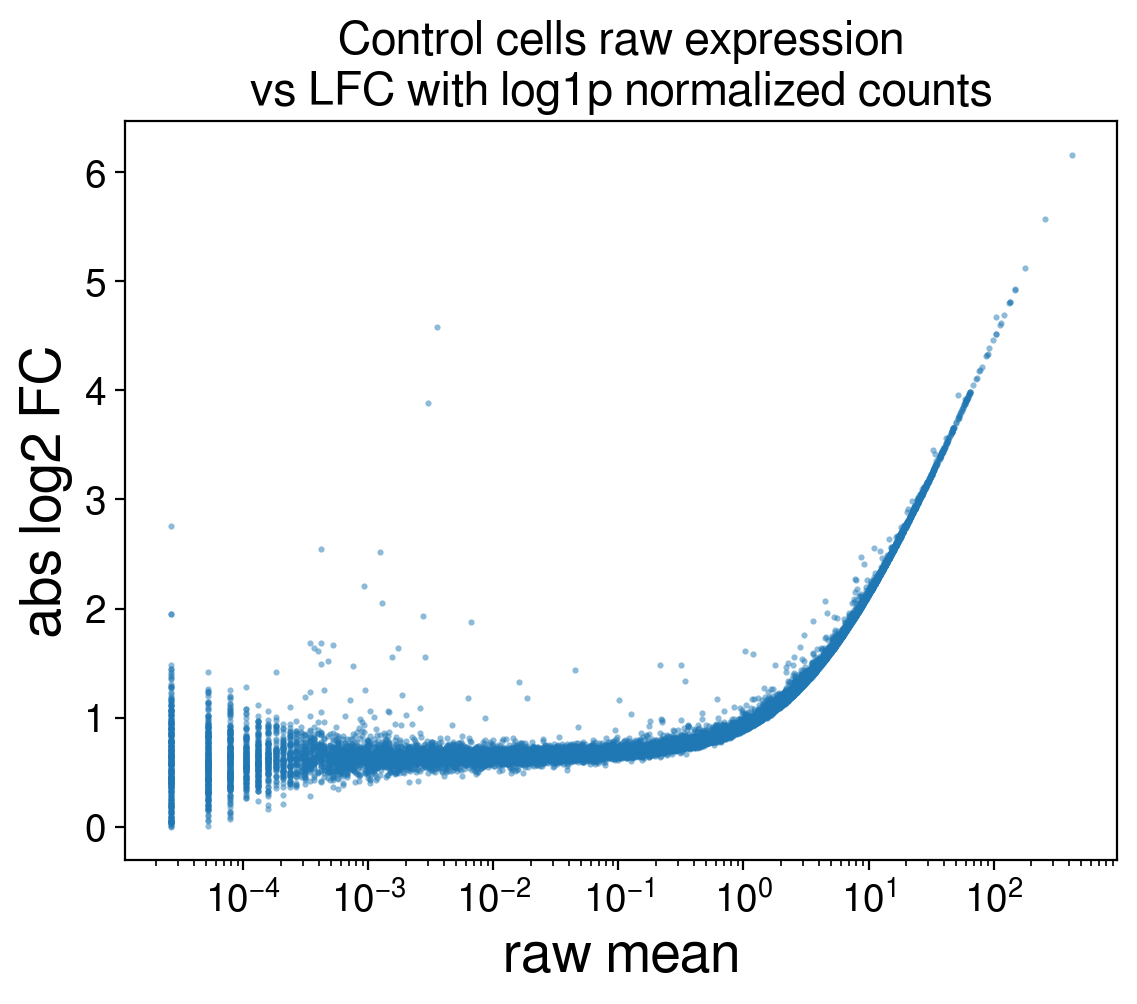
Thus, this approach reproduces the behaviour of DEGs I was exploiting: prioritizing highly expressed genes as DEGs.
While the VCC team updated the cell-eval package to prevent submissions with inconsistent normalization between perturbed and control cells (commit f2519bf), I realized there is another way to accomplish the same result. I could make all the control cell expression values near 0 and use exactly the mean expression of the control cells for all perturbation predictions (baseline). This makes highly expressed genes have a high LFC (proportional to its expression value), and circumvents cell-eval’s checks for reasonable count values. It comes with the additional benefit of preserving the baseline MAE and PDS scores.
When I figured this out, the competition had already begun final submissions, so I could not verify this on their validation set. However, when testing on the training data (I am only using the mean of the non-targeting controls), I get a score of 10.19 (0.32/0.52/0.026).
Side Note on Earlier Submissions
By Harlan Stevens
Before looking at using nUMIs/nCells and DEG-expression relationships in the training set, we tried a few random methods. We’re including these early tests here more for the sake of completeness than any grand insight into making a virtual cell model. We ran the results below before the PDS score fix, so take their raw scores with a grain of salt. That said, our PDS scores were relatively, so we expect the relative ranking of these approaches to be mainly the same.
Surprisingly, the most effective method we tried early on was simply validation gene names onto the closest gene names in the training set (e.g., if ZNF108 appeared in validation, we substituted ZNF204 or whichever was most similar alphabetically). Only about 50% of the genes had a plausible match, so for the rest we defaulted to the control cells. This super simple approach gave a 4.78 (0.164/0.55/0.52). In principle, this is similar to the nearest neighbor method used with nCell and nUMI that gave a 9.4 before the PDS score fix, except here not every perturbation had a match. This suggested that gene names encode some weak biological prior, leading us to try more biologically-informed ways of mapping validation perturbations to their training-set neighbors.
Weirdly, this didn’t really work. Using GO-term similarity gave a 4.46 (0.15/0.56/0.51) and using the presumably better gene embeddings from the PRESAGE model [3]) gave an even worse result 3.40 (0.196/0.51/0.47). We didn’t spend too long digging into why these methods did worse than just the gene-name, but one idea is that we forced every perturbation to have a nearest-neighbor with these, potentially forcing relationships where none exist, while with gene-name similarity, we defaulted to the control cells if there was no good mapping.
We also attempted to improve these mappings by denoise the training data using a method inspired by a recent Perturb-seq paper on overloading guides/cells for cheaper experiments[4]. Yao et al. showed that projecting gene expression into gene-program space and then back again (implemented in FR-Perturb) could act as a denoising step. Unfortunately, running this on the VCC data made the results consistently worse.
Finally, we tried a method based on the perturbMean baseline from the STATE paper [5]), which maps perturbation LFCs from an external dataset onto a cell line of interest. We directly transferred the LFCs from Replogle GWPS in R593 cells [6] for the 47/50 overlapping validation genes onto the control cells. This gave a 4.31 (0.22/0.52/0.47). We tried to improve this by regressing on the overlapping perturbations between the training set and Replogle, and that gave a slightly better 4.54 (0.23/0.51/0.47). Interestingly, these external-dataset mappings tended to achieve better DEG recovery, while the training-set gene-name–based mapping gave better PDS. But since all of these were run before the scoring fix, it’s hard to interpret the tradeoff with much confidence.
Some thoughts
Of course, making all control cells near 0 is not a real solution, and other teams did a much better job at hacking. Still, the experience has helped us appreciate some fundamental issues with the metrics and the overall design of the challenge, as many others have already pointed out online or in discussion.
The definition of a DEG in the VCC is a major weakness. Most bioinformatic analyses utilize a log fold change cut off, which would ameliorate some of the bias towards high expressed genes. While PDS and MAE were supposed to offset the gains from targeted approaches to maximize DES, the construction of the challenge made it possible to still do so without penalty.
If we are using MAE as a component (though arguably, VCC is not due to the nature of the score and clipping at baseline), why can participants submit their own control cells?
Generally, there were too many degrees of freedom that the scores could depend on. I could submit any number of cells per perturbation, or any number of control cells.
While there is plenty of discussion of what are better metrics to evaluate this perturbation prediction task, I think the ambiguity of this problem reflects a deeper issue. We humans do not really have any real intuition on what an 18k-dimensional vector should “look like.” Thus, we have to come up with heuristics to guess at the quality and accuracy of generated single cell data. Distance in these expression spaces do not have consistent meaning, so why are we evaluating models in this space? In comparison, the CASP protein folding competition RMSD metric directly means something biologically relevant (the actual position of atoms in space).
I think a more rational metric should be based in a lower dimensional space. It could be in predicting the activation of covarying gene programs, or cell morphological or functional features. Such a design would be more in line with the success of many models in clinical applications, such as predicting patient survival or therapy outcomes. This idea is also consistent with training models using loss functions in a lower dimensional embedding space (JEPA [7]) or contrastive loss [8].
I’m looking forward to learning what other clever methods teams have come up with to “hack” this challenge, and I happy to share the other ideas we tried that were not discussed here. While hacking is not strictly “in spirit” of the actual goal, it is an important exercise that helps everyone understand the true nature of the data and the proxy tasks we are gauging success on. For a first iteration of the competition, the VCC has definitely succeeded in generating a lot of excitement in the broader ML/bio community that sets the stage for the future of virtual cell models.
Acknowledgements
Thank you to my classmates Harlan Stevens and Arya Rao for your insights and suggestions. Also a big thank you to my PI Fei Chen for letting me take some time to do this challenge; Dan Lesman, Dora Li, Sandeep Kambhampati, and Julie Laffy for our many discussions; and the rest of the Chen lab for their feedback and support.
References
Roohani, Y. H. et al. Virtual Cell Challenge: Toward a Turing test for the virtual cell. Cell 188, 3370–3374 (2025).
gmdbioinformatics. Arc Virtual Cell Challenge: Has the Evaluation collapsed? Genomics, Machine Learning, and Data for Bioinformatics https://gmdbioinformatics.substack.com/p/arc-virtual-cell-challenge-has-the (2025).
Littman, R. et al. Gene-embedding-based prediction and functional evaluation of perturbation expression responses with PRESAGE. Preprint at https://doi.org/10.1101/2025.06.26.661135 (2025)
Yao, D., Binan, L., Bezney, J. et al. Scalable genetic screening for regulatory circuits using compressed Perturb-seq. Nat Biotechnol 42, 1282–1295 (2024).
Adduri, A. et al. Predicting cellular responses to perturbation across diverse contexts with State. Preprint at https://doi.org/10.1101/2025.06.26.661135 (2025).
Replogle, J. et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022).
Assran, M. et al. Self-Supervised Learning from Images with a Joint-Embedding Predictive Architecture. Preprint at https://doi.org/10.48550/arXiv.2301.08243 (2023).
Bahrami, M., Tejada-Lapuerta, A., Becker, S., G, F. S. H. & Theis, F. J. scConcept: Contrastive pretraining for technology-agnostic single-cell representations beyond reconstruction. 2025.10.14.682419 Preprint at https://doi.org/10.1101/2025.10.14.682419 (2025).
 | A guest post by
|
Great post! I missed most of the discussion of this competition, so I wanted to ask: are there any interesting and useful for science solutions on top or hacking won?